“Don’t test me!”— that’s a warning that I, along with my younger brother, Erik, heard more than once from our parents while growing up. The warning would come up from time to time—but particularly during our teenage years—when teenagers do what they sometimes do: challenge authority or just forget some responsibility. (Of course, I now recognize and understand this with two teenage boys in the hopper and a tween daughter not too far away from true teenage status.) In any case, hearing “Don’t test me!” from my parents meant that they were serious and more than willing to impose a penalty for the infraction identified if it happened. FDA has thrown down a similar gauntlet in the 180-day exclusivity arena.
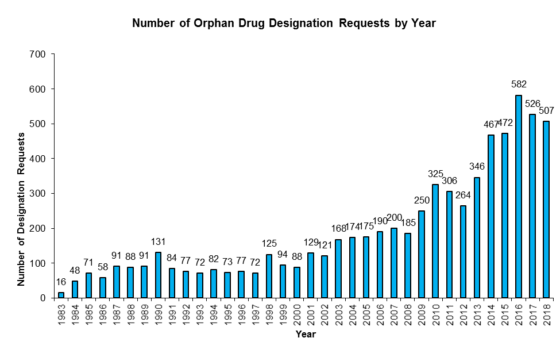
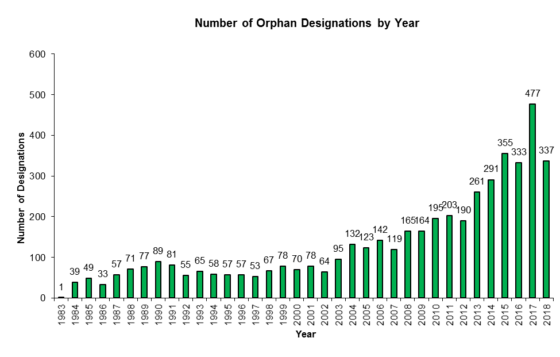
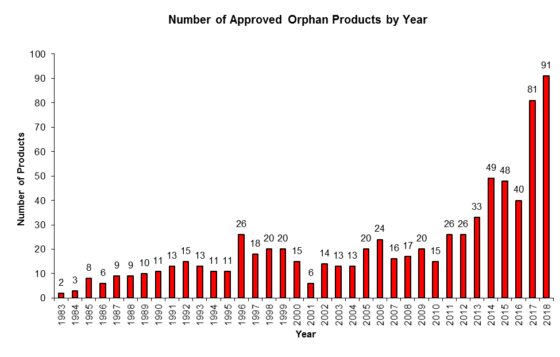
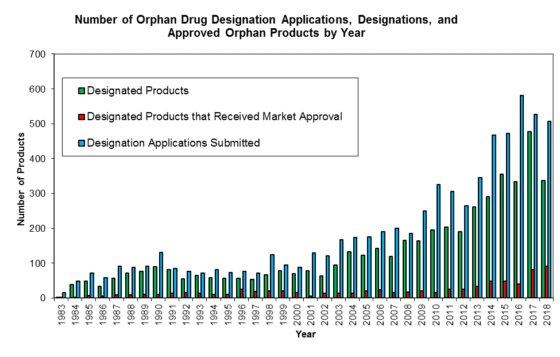
Over the years here at the FDA Law Blog when putting together and updating our popular 180-Day Exclusivity Tracker (which tracks all grants and forfeitures of Paragraph IV 180-day exclusivity and Competitive Generic Therapy (“CGT”) 180-day exclusivity) we’ve found it a bit frustrating that the Orange Book does not always capture the running of a period of 180-day exclusivity. And, in our experience, that’s happened because, despite boilerplate ANDA approval letter language that a company “[p]lease submit a correspondence to this ANDA informing the Agency of the date you begin commercial marketing,” some generic drug manufacturers have failed to notify FDA about the date of first commercial marketing of a drug product so that the Agency can update the Orange Book with the appropriate “PC” (Patent Challenge) exclusivity expiration date. (Failure to notify has not been an issue with CGT 180-day exclusivity, but it’s worth noting that notice timing is important—see our previous post here.)
Back in October 2016 when FDA updated the Agency’s Hatch-Waxman regulations to incorporate some of the changes made by the 2003 Medicare Modernization Act (see our previous post here), the Agency added a new regulation: 21 C.F.R. § 314.107(c)(2). That regulation, which concerns the timing of approval of “subsequent Paragraph IV ANDA” (i.e., an ANDA potentially blocked from final approval because of a first applicant’s eligibility for, or the running of, 180-day exclusivity) states:
A first applicant must submit correspondence to its ANDA notifying FDA within 30 days of the date of its first commercial marketing of its drug product or the reference listed drug. If an applicant does not notify FDA, as required in this paragraph (c)(2), of this date, the date of first commercial marketing will be deemed to be the date of the drug product’s approval.
At the time FDA issued the regulation, we scoffed at it a bit. Would FDA really carry through with this threat? After all, the regulation right below it—“(3) If FDA concludes that a first applicant is not actively pursuing approval of its ANDA, FDA may immediately approve an ANDA(s) of a subsequent applicant(s) if the ANDA(s) is otherwise eligible for approval”—has been on the books for decades and FDA has never exercised that authority (see our previous post here about that regulation and the recently introduced BLOCKING Act). So why would FDA decide to exercise its authority here?
Well, it turns out that despite going soft on the “active pursuit” regulation at 21 C.F.R. § 314.107(c)(3) for so many years, FDA almost immediately took a hard line on the new “deemed triggered” regulation at 21 C.F.R. § 314.107(c)(2). In fact, FDA’s stance has punished some folks who perhaps should not have been punished in the first place.
The instances in which FDA has applied 21 C.F.R. § 314.107(c)(2) are difficult to find and require some hunting around for facts. (And the lack of ANDA approval letters posted on Drugs@FDA does not help.) In fact, the instances—of which there may be a handful—don’t appear to be documented at FDA in any exclusivity memoranda. They just happen.
The first case we noticed occurred with the publication of the July 2017 Orange Book Cumulative Supplement. There, FDA updated the Orange Book to reflect a period of 180-day exclusivity for ANDA 204065 for Desvenlafaxine Succinate Extended-release Tablets, 25 mg, 50 mg, and 100 mg. That ANDA was approved on July 29, 2016. The PC expiration date assigned to the 25 mg strength under ANDA 204065 was January 25, 2017, while the date assigned to the 50 mg and 100 mg strengths approved under ANDA 204065 was August 28, 2017.
At first glance, the different PC expiration dates don’t seem terribly out of order. After all, there are many instances of staggered 180-day exclusivity expiration dates based on different commercial marketing dates. But what stood out here was the date: January 25, 2017. First, is it the only January 25, 2017 date listed in the Orange Book for any of the shared first applicants for any Desvenlafaxine Succinate Extended-release Tablets drug products. The shared first applicants for the 50 mg and 100 mg strengths—ANDA 204003, ANDA 204028, ANDA 204082, ANDA 204083, ANDA 204095, and ANDA 204172—are, along with ANDA 204065, identified with a August 28, 2017 180-day exclusivity expiration date. Second, the “Marketing Start Date” identified on DailyMed for the 25 mg strength approved under ANDA 204065 is March 1, 2017, and that date bears no 180-day relation to January 25, 2017. Third, January 25, 2017 is exactly 180 days from July 29, 2016—the date of approval of ANDA 204065.
The second case we noticed also occurred with the publication of the July 2017 Orange Book Cumulative Supplement. There, FDA updated the Orange Book to reflect a period of 180-day exclusivity for ANDA 204029 for Clofarabine 20 mg/mL (1 mg/mL). That ANDA was approved on May 9, 2017. The PC expiration date assigned to the ANDA was November 5, 2017, which is 180 days after the May 9, 2017. According to DailyMed, the “Marketing Start Date” for Clofarabine 20 mg/mL (1 mg/mL) approved under ANDA 204029 was May 10, 2017, which would have resulted in a November 6, 2017 180-day exclusivity expiration date.
The third case we’ve been able to uncover is a doozy and shows how FDA’s interpretation and application of the “deemed triggered” regulation at 21 C.F.R. § 314.107(c)(2) can have unintended consequences for other ANDA applicants. But just as my parents would say “Too bad!” in response to my brother any his cry of “That’s not fair, Mom/Dad!” when being roped into punishment for one of my infractions, FDA’s response—insofar as how the Agency has interpreted and applied the regulation—seems to be of a similar flavor.
In the January 2019 Orange Book Cumulative Supplement, FDA updated the Orange Book to reflect a period of 180-day exclusivity for ANDA 208327 for Abiraterone Acetate Tablets, 250 mg, that expires on April 29, 2019. FDA approved ANDA 208327 on January 7, 2019, and, according to DailyMed, the “Marketing Start Date” was January 7, 2019. So what gives? Based on the timing of approval of ANDA 208327 and the publication of the January Orange Book Cumulative Supplement, something does not add up.
In this case, there were multiple first applicants eligible for 180-day exclusivity for Abiraterone Acetate Tablets, 250 mg. Subtracting 180 days from the April 29, 2019 exclusivity expiration date identified by FDA yields October 31, 2018. It just so happens that FDA approved four ANDAs on that date: ANDA 208453, ANDA 208339, ANDA 208446, and ANDA 208432. According to DailyMed, product was not marketed under those ANDAs until November 21, 2018 (ANDA 208446 and ANDA 208432) and November 23, 2018 (ANDA 208453 and ANDA 208339), so one might expect 180-day exclusivity to expire on May 20, 2019, which is 180 days from November 21, 2018. But one of those first applicants apparently failed to timely notify FDA of commercial marketing under their ANDA. That caused FDA to set the shared 180-day exclusivity period for all first applicants to 180 days after that applicants October 31, 2018 ANDA approval date. In others words, one slacker first applicant can spoil 180-day exclusivity for all diligent first applicants. That’s an interpretation and application of the “deemed triggered” regulation that’s going to sit well with folks and that might just come under some scrutiny.